|

CFTR Modulation: Today and Tomorrow
In this Issue...
This issue departs from the usual format, as Dr. Claire Wainwright and Dr. Tonia Douglas from Lady Cilento Children's Hospital in Brisbane, Australia review the evidence describing CFTR modulator therapies in current use, new CFTR modulator therapies in the development pipeline, and approaches to optimizing CFTR modulator therapy.
|
|
|
COMPLETE THE
POST-TEST
Click on link to download instructions for the post-test and evaluation


|
|
|
|
|
LEARNING OBJECTIVES
|
|
- Summarize the recent clinical trials of CFTR modulators and their use for different CFTR mutations in genotyped patients with cystic fibrosis.
- Explain optimizing the use of CFTR modulators to maximize the health care benefits.
- Discuss the challenges and future directions for the development of CFTR modulators for all people with cystic fibrosis.
|
|
GUEST AUTHORS OF THE MONTH
|
|
|
|
Commentary & Reviews
|
|
Guest Faculty Disclosure
Dr. Claire Wainwright has indicated that she has received honoraria from Novartis Pharmaceutics, Vertex Pharmaceuticals, and Vertex Pharmaceuticals (Australia) and served as a consultant/advisor and investigator in clinical trials for Vertex Pharmaceuticals.
Dr. Tonia Douglas has indicated that she has no financial interests or relationships with a commercial entity whose products or services are relevant to the content of this presentation.
Unlabeled/Unapproved uses
Dr. Claire Wainwright and Dr. Tonia Douglas have indicated that there will be references to unlabeled or unapproved use of ataluren.
|

|
Claire Wainwright, MBBS, MRCP, FRACP, MD
Professor
University of Queensland
Lady Cilento Children's Hospital
Brisbane, Australia
|

|
Tonia Douglas, MBChB, FRACP, MD
Senior Clinical Lecturer
University of Queensland
Lady Cilento Children's Hospital
Brisbane, Australia
|
|
|
IN THIS ISSUE
|
|
COMMENTARY
Clinical Trials of Ivacaftor
Clinical Trials of Lumacaftor and Lumacaftor/Ivacaftor Combination Therapy
Clinical Trials of Ataluren (PTC124)
Optimizing the Clinical Use of CFTR Modulator Therapy
Pipeline Therapies
KEY TAKEAWAYS
|
|
Program Directors
Peter J. Mogayzel, Jr., MD, PhD
Director, Cystic Fibrosis Center
Professor of Pediatrics
The Johns Hopkins University
Baltimore, Maryland
Noah Lechtzin, MD
Director, Adult Cystic Fibrosis Program
Associate Professor of Medicine
The Johns Hopkins University
Baltimore, Maryland
Suzanne Sullivan, RN, BSN
Senior Clinical Nurse
Johns Hopkins Hospital
Baltimore, Maryland
|
|
|


|
|
COMPLETE THE
POST-TEST
Click on link to download instructions for the post-test and evaluation
|
|
|
|
|
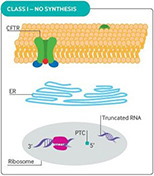
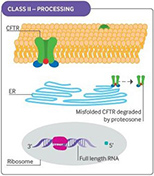
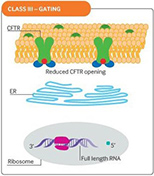
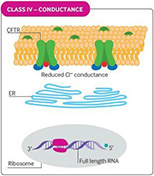
Cystic fibrosis (CF) is caused by defective or deficient cystic fibrosis transmembrane conductance regulator (CFTR) protein, which functions as an anion channel, predominantly in epithelial cell membranes. Since the CFTR gene was discovered in 1989,1 over 2000 different CFTR mutations have been reported, although not all of them have functional consequences.2 Different classes of CFTR mutation have been described that result in inadequate CFTR protein production, deficient CFTR function at the epithelial cell membrane, or a combination of these abnormalities (see Fig 1). Deficient function may arise from reduced open probability of the CFTR channel (gating), abnormal conductance of the channel, or both. The prevalence of CFTR mutations differs among populations, but the most common CFTR mutation is the Phe508del (F508del) mutation, with around 45% of patients homozygous for this class II mutation worldwide.3
The advent of CFTR modulator therapy in the last decade has revolutionized the approach to CF treatment by targeting the underlying CFTR defect. The rapidity with which these novel agents have progressed from preclinical trials to clinical use and the expanding research into more potent drugs targeting a broader range of mutation classes are at once exciting and challenging.
The first CFTR modulator approved for clinical use was ivacaftor; a CFTR potentiator that increases the open probability (gating) of the CFTR channel and targets class III CFTR gating mutations. The most common gating mutation is the G511D (Gly551Asp) mutation, accounting for approximately 4% of CFTR alleles and associated with adequate CFTR protein production but no CFTR function. Two landmark phase 3 trials of ivacaftor in patients carrying at least one copy of the G511D (Gly551Asp) mutation4,5 demonstrated improvements in lung function and nutritional status and reported a marked reduction in sweat chloride measurement, confirming restoration of CFTR function. These clinical benefits persisted in a two-year open label extension study.6 In 2012 the United States Food and Drug Administration (FDA) approved the use of ivacaftor for patients aged 6 years and older with CF who carry at least one G511D (Gly551Asp) mutation. Since then, approved ivacaftor use has extended to eight other (non-G511D) gating mutations (July 2014);7 to patients carrying the residual function R117H (Arg117His) mutation (December 2014);8 and in March 2015, the FDA reduced the age of access for these patients to age 2 years and older.
Correcting the CFTR abnormalities associated with the Phe508del class II CFTR mutation is a much greater challenge. Therapies targeting the Phe508del mutation must modulate both the CFTR processing defect and the gating defect. In vitro studies of the Phe508del CFTR corrector, lumacaftor, showed increased CFTR9 production, and in combination with ivacaftor10, greater chloride transport than with either drug alone.9 As monotherapy with each of these agents was not found to have clinical benefit11,12, combination therapy with lumacaftor and ivacaftor was studied in two large phase 3 trials of patients homozygous for Phe508del-CFTR. Relatively modest improvements in lung function, nutrition, and pulmonary exacerbations13 were observed. While less impressive than for ivacaftor, the findings are arguably no less clinically significant when the size of the target population is considered with the potential positive implications for long-term morbidity. Combination therapy with lumacaftor and ivacaftor was approved by FDA in July 2015 for patients aged 12 years and older homozygous for Phe508del-CFTR.
There has been less success to date with CFTR modulator therapies for class I CFTR mutations, and the phase 3 trial of ataluren in patients with CF carrying a class I mutation did not result in clear clinical benefit.14
An important challenge for clinicians is how to optimize the use of CFTR modulators in their patients. Considerations include selecting patients on the basis of genotype, ability to adhere to therapy, and severity of disease. Despite the promise of benefit with ivacaftor, adherence may not be much better than is seen for other CF therapies.15 In addition, patients who feel healthier taking CFTR modulators may reduce adherence to other daily therapies, thereby risking health maintenance. Clinical decisions surrounding initiation of treatment in patients with mild or severe lung disease are difficult on the basis of current evidence. Indications for treatment may extend beyond improvement in lung function and nutrition as more evidence emerges regarding additional effects of CFTR modulators in CF, such as mucociliary clearance, airway microbiology16, and glucose homeostasis.17 Maintaining optimal outcomes with CFTR modulator therapies as they become available will require careful assessment of patient CF genotype; the potential for clinical benefit in conjunction with education and support to ensure adherence; and monitoring for adverse events and drug interactions. The ultimate goal of disease prevention has already driven clinicians to start therapies earlier in life and in milder disease. While the potential benefit of this approach is clear, it raises the importance of long term monitoring of therapies introduced in infancy and potentially continuing across the life span.
A major challenge for the future of CFTR modulation is to develop therapies that are effective in all patients; that is, that are not mutation class specific. As novel agents move from preclinical studies to clinical trials18, there is an obligation to broaden the therapeutic repertoire to target the full range of basic defects caused by the different CFTR mutation classes and ensure equitable access to all patients.
Clinical trials provide evidence for the efficacy of new drugs, yet many patient groups are not included. Trials are more challenging in young children, in sicker patients, and in those with rarer CFTR mutations where patient numbers may be too small to power conventional clinical trials. New approaches will be needed to ensure equitable access to new therapies for all patients with CF. As more patients start to use CFTR modulator therapy, the pool of patients naïve to CFTR modulator therapy declines, making placebo controlled trials less possible.
Alternatives to the typical randomized controlled design will be needed in future clinical trials, including protocols that accommodate small samples using more sensitive outcome measures of effect and employing design approaches that can obtain sufficient meaningful data to enable extension of the findings to larger populations. Comparative effectiveness studies will likely be used, and patients, health care providers, researchers, and pharmaceutical companies will need to work together closely to realize the potential for these new medicines for people with CF.
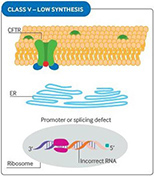
Fig 1. CFTR mutational classes and molecular consequences.
 |
 |
 |
| Class I mutations result in unstable truncated RNA and no synthesis of the CFTR protein. |
Class II mutations cause CFTR processing defects owing to misfolding of CFTR and degradation by the proteasome. |
Class III mutations cause reduced CFTR channel opening owing to defective channel gating or regulation. |
 |
 |
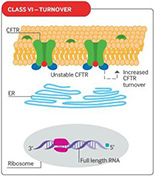 |
| Class IV mutations cause reduced chloride conductance owing to defects within the CFTR channel. |
Class V mutations lead to reduced synthesis of CFTR owing to splicing defects. |
Class VI mutations result in reduced CFTR stability at the cell surface and hence increased CFTR turnover. |
Reproduced with permission from Quon BS,Rowe SM. New and emerging targeted therapies for cystic fibrosis. (Fig 3). BMJ. 2016 Mar 30;352:i859.
The size of the inner dark circle of the CFTR channel at the apical surface reflects the extent of channel opening or conductance.
CFTR = cystic fibrosis transmembrane conductance regulator; ER = endoplasmic reticulum; PTC = premature termination codon.
References:
- Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989;245:1073-1080.
- Sosnay PR, Siklosi KR, Van Goor F, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet. 2013;45:1160-1167.
- CFTR2 website. The clinical and functional translation of CFTR (CFTR2). 2011. Available at http://cftr2.org.)
- Ramsey BW, Davies J, McElvaney NG, et al; for the VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663-1672.
- Davies JC, Wainwright CE, Canny GJ, et al; on behalf of the VX08-770-103 (ENVISION) Study Group. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187:1219-1225.
- McKone EF, Borowitz D, Drevinek P, et al; VX08-770-105 (PERSIST) Study Group. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the G511D (Gly551Asp)-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir Med. 2014;2:902-910.
- De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13:674-680.
- Moss RB, Flume PA, Elborn JS, et al; on behalf of the VX11-770-110 (KONDUCT) Study Group. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3:524-533.
- Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108:18843-18848.
- Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106:18825-18830.
- Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012;67:12-18.
- Flume PA, Liou TG, Borowitz DS, et al; for the VX 08-770-104 Study Group. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012;142:718-724.
- Wainwright CE, Elborn JS, Ramsey BW, et al; for the TRAFFIC and TRANSPORT Study Groups. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220-231.
- Kerem E, Konstan MW, De Boeck K, et al; Cystic Fibrosis Ataluren Study Group. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2:539-547.
- Siracusa CM, Ryan J, Burns L, et al. Electronic monitoring reveals highly variable adherence patterns in patients prescribed ivacaftor. J Cyst Fibros. 2015;14:621-626.
- Rowe SM, Heltshe SL, Gonska T, et al; GOAL Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. 2014;190:175-184.
- Bellin MD, Laguna T, Leschyshyn J, et al. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatr Diabetes 2013;14:417-421.
- Cystic Fibrosis Foundation Website. Drug Development Pipeline page. Available at: https://tools.cff.org/research/drugdevelopmentpipeline/. Accessed April 20 2016.
Back to top
|
|
Clinical Trials of Ivacaftor
|
|
|
COMPLETE THE
POST-TEST
Click on link to download instructions for the post-test and evaluation
|
|
|
|
|
Ivacaftor is the most clinically effective CFTR modulator to date. This small-molecule potentiator facilitates CFTR function at the cell membrane by increasing chloride ion conduction, although the precise mechanism of action is unclear. Ivacaftor has demonstrated clinical efficacy in patients with class III gating mutations and in combination with a CFTR corrector in patients with class II mutations.
FDA approval for ivacaftor was based on the findings of two landmark phase 3 randomized, double blind, placebo controlled clinical trials: STRIVE and ENVISION.1,2 The STRIVE trial randomized 161 patients carrying one or more G511D (Gly551Asp) CFTR mutations, aged 12-53 years (FEV1 40%-90%) to 150 mg ivacaftor twice daily or placebo for 48 weeks. The primary endpoint was the change in percent predicted FEV1 from baseline, with secondary endpoints including change in body weight, pulmonary exacerbations, and sweat chloride, and change in the respiratory domain of the Cystic Fibrosis Quality of Life- revised tool (CFQ-R). At 24 weeks approximately three-quarters of subjects demonstrated an increase in FEV1 of 5% or more that persisted to 48 weeks. The improvement in lung function was rapid and apparent by two weeks of ivacaftor treatment. At 48 weeks, patients randomized to ivacaftor demonstrated a 10.5% increase in percent predicted FEV1 compared to placebo (P < .001); had significantly fewer pulmonary exacerbations (67% vs 41% exacerbation free; hazard ratio 0.455, P = .001); a reduction in sweat chloride (-47.9 mmol/LP < .001); and increase in body weight (3.1 kg vs 0.4kg, P = .001). Quality of life scores in the respiratory domain were 8.6 percentage points higher in the ivacaftor group at 48 weeks compared to placebo (P < .001). There were no significant differences in safety and tolerability between the ivacaftor and placebo groups, with fewer serious adverse events related to a reduction in pulmonary exacerbations observed with ivacaftor compared with placebo.
Efficacy of ivacaftor in children was confirmed in the ENVISION trial that randomized 52 children aged 6-11 years, carrying one or more G511D (Gly551Asp) CFTR mutations, to 150 mg ivacaftor or placebo for 48 weeks. Using the same framework and endpoints as in the STRIVE trial, ENVISION demonstrated comparable improvements to STRIVE in lung function at 24 weeks (change in percent predicted FEV1 from baseline: ivacaftor 12.6% vs placebo 0.1%, P < .001) that persisted to 48 weeks; reduction in sweat chloride (ivacaftor -54.3 mmol/L vs placebo -1.2 mmol/L, P < .001); weight gain (3.7 kg vs 1.8 kg, P < .001 at 24 weeks, sustained to 48 weeks in the ivacaftor group); and a 6.1-point increase in CFQ-R respiratory domain (P = .109). The rate of pulmonary exacerbations was too low for meaningful analysis (ivacaftor 4 events vs placebo 3 events).
Improvement in lung function was greatest among patients with lower lung function, although significant increases were also observed in those with near normal values. Overall adherence to study treatment was high (~ 90% STRIVE, ~ 95% ENVISION).
Longer-term clinical efficacy and safety data were obtained in the 2014 PERSIST study, a 96-week open-label extension trial of patients who had completed STRIVE or ENVISION.3 The study enrolled 144 adults (STRIVE) and 48 children (ENVISION) to receive 96 weeks of ivacaftor 150 mg BID, evaluating the same clinical end points. The individuals enrolled from the STRIVE/ENVISION placebo groups who began ivacaftor in PERSIST showed improvements in lung function, body weight, and CFQ-R scores comparable to those observed in the treatment arms of the prior studies, and these improvements were sustained throughout the 96 week trial period. Annualized pulmonary exacerbation rates decreased following their 48 weeks of ivacaftor treatment. In STRIVE and ENVISION subjects previously treated with ivacaftor, the improvements in clinical outcomes observed were sustained throughout the additional 96 weeks of ivacaftor treatment (total 144 weeks’ treatment). No new clinical safety concerns were raised during the extension study, and adverse events were comparable in nature and frequency to those observed in STRIVE and ENVISION, with pulmonary exacerbations, upper airway symptoms, and mild gastrointestinal symptoms being the most common.
Regulatory body approval for the expansion of the indication for ivacaftor to individuals³ aged 2 years and older, and to those carrying one of eight other gating mutations was obtained based on the data from the KONNECTION (2014)4 and KIWI (2016) studies.5 De Boeck and colleagues4 demonstrated clinical efficacy of ivacaftor in 39 patients (³ 6 years) carrying non-G511D (Gly551Asp) CFTR gating mutations (G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, and G1349D), in an eight-week, double blind, crossover study, followed by a 16 week open-label extension period. The primary outcome measure was the absolute change in percent predicted FEV1 at eight weeks and 24 weeks, with secondary endpoints as change from baseline in body mass index (BMI), CFQ-R respiratory domain, and sweat chloride. At eight weeks ivacaftor was associated with a significant increase in percent predicted FEV1 (7.5% vs -3.2%, P > .0001), BMI z score (0.24 vs -0.04 points, P = .001), and CFQ-R (8.9 points vs -0.7, P = .0004) compared to placebo. Sweat chloride levels significantly decreased in the treatment group (-52.3 mmol/L vs -3.1 mmol/L, P < .0001). These observations were sustained in the 16-week extension period, with significant and impressive improvements from baseline in percent predicted FEV1, BMI, CFQ-R and sweat chloride at 24 weeks.
Davies and colleagues5 conducted the first trial of ivacaftor monotherapy in young children aged 2-5 years with one or more G551D (Gly551Asp) CFTR mutations, in an open-label, single arm, two-part study. The primary outcome was safety and pharmacokinetic data, with secondary endpoints of change in sweat chloride, weight, BMI, and height reported. Part A was a four-day safety and pharmacodynamics study of ivacaftor, 50 mg (subjects < 14 kg) or 75 mg (³ 14 kg) given twice daily, in nine subjects. Results confirmed area under the curve (AUC) values comparable to those in adults and acceptable short-term safety for continuation to Part B. Part B was a 24-week open label study of 34 children treated with ivacaftor that demonstrated significant reduction in sweat chloride levels from baseline (mean change -46.9 mmol/L, P < .0001), a 0.2 point (P = .001) and 0.4 point (P < .0001) increase in weight and BMI z scores respectively. Observations of improved pancreatic function (increased fecal elastase-1 concentration) in some children did not reach statistical significance but are nevertheless intriguing. The safety profile was similar to that observed in adults and older children, with the most common adverse effect being cough (56%), vomiting (29%), and upper respiratory symptoms.
The finding of elevated transaminases in 15% of the cohort raised concerns about the potential for hepatic injury in young children receiving ivacaftor. The results of an 24-week open extension study found that five (15%) of 34 children enrolled in KIWI reported elevated liver enzymes greater than eight times the upper limit of normal. Of these, four children had study drug interruption, and one child discontinued ivacaftor. The effects on sweat chloride and nutritional outcomes were sustained throughout the study, and overall the safety profile appeared to be otherwise comparable to those in adults and older children. These data suggest ivacaftor is palatable, safe, and well tolerated in preschool children with data to support clinical efficacy, although close monitoring of liver enzymes is required.
The clinical effect of ivacaftor in residual function CFTR mutations (class IV) has been examined in a phase 3 randomized, controlled, double-blind trial of 69 patients³ 6 years with the R117H (Arg117His) CFTR mutation in association with a second, nonfunctional protein mutation (KONDUCT, 2015).6 The absolute change in percent predicted FEV1 from baseline was evaluated over a 24-week period, followed by a 12- week open-label extension study. No significant difference in percent predicted FEV1 from baseline (2.6% vs 0.5%, P = .2) or BMI (0.49 vs 0.23 kg/m2, P = .78) was observed with ivacaftor compared to placebo at 24 weeks, although there were significant improvements in CFQ-R respiratory domain (8.4 points, P = .009) and reduction in sweat chloride (-24 mmol/L, P < .001). Of note was the high mean baseline values of lung function in patients in the 6-11-year age group that may have masked a treatment effect.
A subgroup analysis of patients³ 18 years and older (with correspondingly lower mean baseline FEV1) yielded more promising results, with a 5% point increase in FEV1 from baseline with ivacaftor compared to placebo, (4.5% vs - 0.5%, P = .01). No new safety concerns were evident in the trial or in the extension period, and the safety profile was comparable to those in the STRIVE and ENVISION studies.
References:
- Ramsey BW, Davies J, McElvaney NG, et al; VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663-1672.
- Davies JC, Wainwright CE, Canny GJ, et al; VX08-770-103 (ENVISION) Study Group. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187:1219-1225.
- McKone EF, Borowitz D, Drevinek P, et al; VX08-770-105 (PERSIST) Study Group. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir Med. 2014;2:902-910.
- De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13:674-680.
- Davies JC, Cunningham S, Harris WT, et al; KIWI Study Group. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;4:107-115.
- Moss RB, Flume PA, Elborn JS, et al; VX11-770-110 (KONDUCT) Study Group. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3:524-533.
Back to top
|
|
Clinical Trials of Lumacaftor and Lumacaftor/Ivacaftor Combination Therapy
|
|
|
COMPLETE THE
POST-TEST
Click on link to download instructions for the post-test and evaluation
|
|
|
|
|
Lumacaftor (VX-809) is a corrector that augments the correct folding and stability of Phe508del (F508del)-CFTR in the cell. Lumacaftor is a chaperone molecule that has been shown in vitro to increase stable Phe508del-CFTR production six-fold, potentially resulting in greater concentrations of CFTR at the cell membrane.1 However, while lumacaftor may increase the quantity of Phe508del-CFTR, it does not appear to restore function to normal levels. A phase 2a randomized double blind, placebo controlled study of lumacaftor in 89 adults homozygous for Phe508del-CFTR suggested a similar safety profile between placebo and active drug, and a small, dose-dependent reduction in sweat chloride levels with active drug.2
A second trial recruited adult patients homozygous (n = 82) and heterozygous (n = 27) for Phe508del-CFTR, randomized to either lumacaftor monotherapy at different doses or placebo for 28 days, followed by a second 28-day period of combination therapy (lumacaftor 600 mg and ivacaftor 250 mg) or placebo.3 In the homozygous group the greatest increase in lung function was observed in the lumacaftor 600 mg/ivacaftor 250 mg group, with a 6.7% absolute increase in percent predicted FEV1 from baseline (P = .002). Statistically significant decreases in sweat chloride from baseline were observed over the total study period, suggesting some restoration of CFTR function with combination therapy. Adverse events were comparable across the treatment and placebo groups with no new safety concerns emerging. Heterozygous patients experienced a decrease in percent predicted FEV1 of -1.3% compared to -3.7% placebo at 28 days, and those taking combination therapy did not experience significant changes in sweat chloride levels. These data suggested that a different approach is needed for heterozygous patients.
The results of these studies informed two 24-week, phase 3 trials of combination therapy with lumacaftor and ivacaftor in patients homozygous for Phe508del-CFTR: TRAFFIC and TRANSPORT.4 These large, multicenter studies randomized 1108 patients aged 12 years and older to receive either lumacaftor 600 mg or 400 mg in combination with ivacaftor 250 mg, or placebo twice daily. Randomization was stratified according to age, gender, and pulmonary function; the primary endpoint was the absolute change in percent predicted FEV1 from baseline at 24 weeks (calculated as an average of the percent predicted FEV1 at weeks 16 and 24). The trial demonstrated a modest but significant increase in percent predicted FEV1 from baseline that was apparent by two weeks of treatment and was sustained for the trial duration in both active treatment groups. A mean relative treatment difference in percent predicted FEV1 of 4.3%-6.7% (P < .001) between active treatment and placebo groups was demonstrated, with 39%-46% of patients in the treatment groups experiencing a relative improvement in percent predicted FEV1 of 5% or more, compared to 22% of those on placebo. Subgroup analyses demonstrated a beneficial treatment effect in adolescents, patients with severe lung disease, and patients with or without Pseudomonas aeruginosa infection. Pooled analysis demonstrated a 30%-39% reduction in pulmonary exacerbations (P < .001), significantly fewer hospitalizations and use of intravenous antibiotics (P < .003), and a 1% improvement in BMI (P < .001) in the active treatment groups compared to placebo. The safety profile was acceptable.
As in the phase 2 studies, there were adverse events related to dyspnea and chest tightness, but these tended to resolve within the first month of use and with use of a bronchodilator. Some abnormalities of liver enzymes were also seen, and more patients on active treatment ceased study drug compared with placebo, although the numbers of patients with elevation of liver enzymes was similar between active and placebo groups. Overall the study confirmed a more modest but clinically significant benefit of combination lumacaftor-ivacaftor therapy for patients homozygous for Phe508del-CFTR, which affects 45% of the population with cystic fibrosis.
References:
- Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108:18843-18848.
- Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67:12-18.
- Boyle MP, Bell SC, Konstan MW, et al; VX09-809-102 study group. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2:527-538.
- Wainwright CE, Elborn JS, Ramsey BW, et al; for the TRAFFIC and TRANSPORT Study Groups. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220-231.
Back to top
|
|
Clinical Trials of Ataluren (PTC124)
|
|
|
COMPLETE THE
POST-TEST
Click on link to download instructions for the post-test and evaluation
|
|
|
|
|
Ataluren is a CFTR modulator that targets nonsense mutations and overrides the premature stop codon that prevents the production of a complete CFTR protein. It is the only therapy for class I CFTR mutations that has reached phase 3 clinical trials and is currently being evaluated in an ongoing 96-week, open label, extension study (NCT02456103) into the long term safety and efficacy among patients not receiving treatment with inhaled aminoglycosides. The safety and efficacy of ataluren has been assessed in adults and children (> 6 years old) in three phase 2 studies.1-3
These studies collectively demonstrated significant improvements in nasal chloride channel activity among patients randomized to ataluren compared to placebo. The results of these studies led to a phase 3 randomized, double-blind, placebo-controlled trial of 238 patients > 6 years, with mean change in FEV1 at 48 weeks as the primary endpoint.4 Results of the trial were disappointing, with a relative overall fall in lung function seen across both active and placebo groups (change from baseline in percent predicted FEV1 of -2.5% ataluren vs -5.5% placebo, P = .099).
A post hoc analysis of patients not receiving aminoglycosides was more promising, showing a 5.7% relative increase in FEV1 from baseline (P = .015) and significantly fewer pulmonary exacerbations (P = .0061) in the treatment group. These data suggest that ataluren may restore the production of CFTR, but concurrent use of aminoglycosides may inhibit the activity and efficacy of ataluren. As aminoglycosides are a cornerstone of antibiotic therapy in CF, the clinical application of ataluren may be limited. Ataluren was generally well tolerated, although observations of elevated serum creatinine (in 15% of the treatment group vs 1% of placebo) raise some concerns regarding the safety of this agent. Longer-term safety and efficacy data will ultimately determine if ataluren is a suitable future therapeutic agent in CF.
References:
- Kerem E, Hirawat S, Armoni S, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372:719-727.
- Sermet-Gaudelus I, Boeck KD, Casimir GJ, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med. 2010;182:1262-1272.
- Wilschanski M, Miller LL, Shoseyov D, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J. 2011;38:59-69.
- Kerem E, Konstan MW, De Boeck K, et al; Cystic Fibrosis Ataluren Study Group. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2:539-547.
Back to top
|
|
Optimizing the Clinical Use of CFTR Modulator Therapy
|
|
|
COMPLETE THE
POST-TEST
Click on link to download instructions for the post-test and evaluation
|
|
|
|
|
The studies reviewed above establish the groundwork of evidence on which to base the use of CFTR modulators in clinical practice. We can appreciate which type of patients are likely to benefit from available CFTR modulators, how they will benefit, and the magnitude of the effects. However, optimizing the clinical application of these drugs requires careful consideration.
Real world use of therapies may yield less impressive or different results in patients1 for a number of reasons; perhaps most important of these is adherence. Adherence to complex therapeutic CF regimens is a huge challenge for patients outside the intensely supportive environment of a clinical trial. Ivacaftor and lumacaftor/ivacaftor combination require twice daily oral administration with fatty food to maximize absorption and bioavailability and will be prescribed under the premise that patients continue standard/usual CF care. Little data is available to guide physicians or patients in how to best rationalize or manage the ever increasing therapeutic burden in CF, and adherence to these exciting new therapies may be less than optimal.
A small, single-center study electronically monitored adherence to ivacaftor in 12 patients (6-48 years) over a mean of 118 days and found that adherence decreased with time and was poor overall at 61% (SD 28%), with a mean time between doses of 16.9 hours (IQR 13.9-24.1 hours). Patients overestimated their adherence (100% by self report vs 84% from prescription refill history and 61% by electronic monitoring)2, and therefore worryingly, possibly 20% of drug may be wasted despite prescription refill. Careful assessment, education, and shared decision-making among health care professionals, patients, and their families will be needed to optimize the outcomes from CFTR modulators.
Optimizing the clinical application of CFTR modulators requires clinicians to think beyond the anticipated benefits in nutrition and lung function. Future considerations for use may include beneficial effects of CFTR modulator therapies on airway microbiology1 and glucose homeostasis.3 Ivacaftor has been associated with a reduction in the percentage of patients with Pseudomonas aeruginosa cultured in sputum (18.8% fewer P = .003)1, although inflammatory parameters and the bacterial load of P. aeruginosa in positive sputum cultures did not change. Cystic fibrosis-related diabetes (CFRD) is a common complication associated with significant morbidity, and understanding is growing that insulin secretion is affected by abnormal CFTR function.3 Enhancing CFTR function may improve glucose tolerance and reduce CFRD. A small pilot study in five children with a G511D CFTR mutation demonstrated improved insulin secretion following glucose challenge after one month of ivacaftor therapy.4 These intriguing observations require further study in larger populations but suggest a promising role for CFTR modulator therapy.
Optimizing clinical use of CFTR modulators means equitable access to therapy for all people with CF. Can we extend the use of these therapies to the very young, those with mild lung disease (FEV1 > 100%), or severe disease (FEV1 < 40% predicted) when trials to date have largely excluded these groups of patients? The usual clinical outcome measure of FEV1 in clinical trials is an insensitive endpoint in mild lung disease. Therefore, the treatment effect of CFTR modulators may be underestimated in patients with preserved lung function. Lung clearance index (LCI) is a measure of ventilation inhomogeneity and appears to be a more sensitive measure in patients with preserved lung function. Davies and coworkers demonstrated statistically significant improvements in LCI (-2.16 P < .0001) as a clinical endpoint in a small crossover trial of 21 patients with a G511D CFTR mutation and mild lung disease (baseline FEV1 > 90%, and LCI > 7.4) who received ivacaftor 150 mg twice daily or placebo with treatment blocks and a washout period of 28 days.5 Future trials are needed using more sensitive end points such as LCI to accurately assess clinical response of this group of patients.6
Patients with severe lung disease are an important and vulnerable group. A small retrospective study of 21 adults with severe lung disease who received ivacaftor on a compassionate use basis showed a significant increase from baseline FEV1 (26.5% to 30.7% predicted P = .01), a decrease in intravenous antibiotic days (23 to 0 days P < .001), and improvement in weight gain (49.8 kg to 51.6 kg P = .006) compared to matched controls.7 Other reports of ivacaftor in severe lung disease report similar findings of increased lung function and weight gain from baseline.8 In the subgroup analysis of 81 patients enrolled in TRAFFIC and TRANSPORT studies with percent predicted FEV1 that fell below 40% between screening and baseline, similar benefits were seen compared with the overall trial population.9 Future trials of CFTR modulators must specifically examine the response to treatment among patients at the extremes of disease spectrum/severity.
A pivotal question for clinicians and researchers is the role of CFTR modulators as a disease modifier in preventing CF lung disease. Lung disease begins very early in life, and chest computed tomography (CT) scan-diagnosed bronchiectasis is established in up to 60%-80% of children by age 5-6 years.10,11 Can CFTR modulator therapy delay or prevent lung disease in young children and infants with CF? Currently evidence is lacking in this area, and clinicians will have to wait for results of well-designed trials in young children that introduce CFTR modulation early in life and measure the effects using robust clinical endpoints. A disease modifying effect of ivacaftor in patients with CF carrying at least one G511D (Gly551Asp) mutation has been suggested in a recent study.12 The magnitude of lung function decline in patients who took part in the clinical trials of ivacaftor and the open label extension study over a three-year period was compared to patients homozygous for Phe508del. Patients were matched at baseline for age, sex, nutrition, FEV1 and chronic therapies. The FEV1 decline was halved in patients treated with ivacaftor, but decline persisted despite treatment with CFTR modulators.
Clearly, optimizing the clinical application of CFTR modulators requires both consideration of the individual and critical evaluation of the available evidence supporting boarder clinical benefits across a wider range of patients.
References:
- Rowe SM, Heltshe SL, Gonska T, et al; GOAL Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. 2014;190:175-184.
- Siracusa CM, Ryan J, Burns L, et al. Electronic monitoring reveals highly variable adherence patterns in patients prescribed ivacaftor. J Cyst Fibros. 2015;14:621-626.
- Koivula FN, McClenaghan NH, Harper AGS, Kelly C. Islet-intrinsic effects of CFTR mutation. Diabetologia 2016: Jul;59(7):1350-1355.
- Bellin MD, Laguna T, Leschyshyn J, et al. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatr Diabetes 2013;14:417-421.
- Davies J, Sheridan H, Bell N, et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir Med. 2013;1:630-638.
- Davies JC, Cunningham S, Harris WT, et al; KIWI Study Group. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;4:107-115.
- Barry PJ, Plant BJ, Nair A, et al. Effects of ivacaftor in patients with cystic fibrosis who carry the G551D mutation and have severe lung disease. Chest 2014;146:152-158.
- Hebestreit H, Sauer-Heilborn A, Fischer R, Kading M, Mainz JG. Effects of ivacaftor on severely ill patients with cystic fibrosis carrying a G551D mutation. J Cyst Fibros. 2013;12:599-603.
- Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220-231.
- Stick SM, Brennan S, Murray C, et al; Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF). Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr. 2009;155:623-628 e1.
- Wainwright CE, Vidmar S, Armstrong DS, et al; ACFBAL Study Investigators. Effect of bronchoalveolar lavage-directed therapy on Pseudomonas aeruginosa infection and structural lung injury in children with cystic fibrosis: a randomized trial. JAMA 2011;306:163-171.
- Sawicki GS, McKone EF, Pasta DJ, et al. Sustained Benefit from Ivacaftor Demonstrated by Combining Clinical Trial and Cystic Fibrosis Patient Registry Data. Am J Respir Crit Care Med. 2015;192:836-842.
Back to top
|
|
Pipeline Therapies
|
|
|
COMPLETE THE
POST-TEST
Click on link to download instructions for the post-test and evaluation
|
|
|
|
|
The demand to broaden the therapeutic repertoire of CFTR modulators to target the full range of basic defects caused by the various mutation classes has resulted in a proliferation of new compounds being developed and assessed in preclinical studies for their potential to modify CFTR function.1,2 The CF Foundation lists active clinical trials of new CFTR modulators3, and at the time of access six new compounds had entered into phase 1-3 trials.
Ataluren (PTC124) and VX-661 have both progressed to phase 3 studies. VX-661 is a class II mutation corrector that functions as a trafficker molecule delivering CFTR to the cell membrane. VX-661 has entered into phase 3 clinical trials in patients with CF. These have included patients who are homozygous for the Phe508del mutation as well as patients with at least one copy of Phe508del mutation. VX-661 in combination with ivacaftor is currently under investigation in patients heterozygous for the Phe508del mutation who have a residual function mutation (NCT02392234) or a gating mutation (NCT 02412111).
Results of phase 2 trials in 128 adults homozygous for Phe508del CFTR mutations found improved benefit/risk profile with VX-661 plus ivacaftor combination compared to lumacaftor/ivacaftor and demonstrated significant increases in FEV1 and reduction in sweat chloride at 28 days.4,5 Results from the phase 3 trials are expected to be available within 12 months.2
A recent press announcement reported that the trials for VX-661 in combination with ivacaftor in patients heterozygous for the Phe508del and with a second mutation that was not likely to respond to the combination therapy (NCT02516410) has been stopped for futility reasons although no concerning safety signals were reported.5
Two “next generation” correctors — VX-152 and VX-400 — are currently being evaluated both as monotherapy and in triple combination with VX-661 plus ivacaftor. Pending the results of the phase 1 dose-response studies, both these triple combinations will move into phase 2 studies.2
A triple combination of novel CFTR potentiators, plus first generation and “new generation” correctors, is in phase 1 and phase 2 trials. Six agents (GLPG1837, GLPG2222, GLPG2451, GLPG2665, GLPG 2737, and GLPG2851), each with a unique mode of action, are being trialed individually and in various combinations to determine safety and efficacy in patients with a range of mutations. In vitro studies of one combination have reported up to a six-fold greater chloride transport than lumacaftor/ivacaftor, and different combinations of these agents may eventually address 80%-90% of mutations in the CF population. Early trial results are expected within 12 months.6-9
Of particular promise are the two phase 2a studies of VX-371, an inhaled epithelial sodium channel (ENaC) inhibitor with potential benefit for all people with CF, regardless of CFTR mutation. One study is evaluating this agent in patients homozygous for Phe508del CFTR mutations who are taking lumacaftor/ivacaftor; the other (CLEAN-CF) is enrolling people with a confirmed diagnosis of CF and any CFTR mutation.2
N9115 is an S-nitrosoglutathione (GSNO)-signaling molecule that improves the stability and quantity of CFTR at the cell membrane. It is the first oral GSNO reductase inhibitor in clinical development. In preclinical studies, N9115 was shown to improve Phe508del-CFTR function, and the agent was evaluated in a phase 1b study. A phase 2 study is now underway evaluating N9115 in combination with lumacaftor/ivacaftor.10,11 An intravenous GSNO reductase inhibitor, N6022, is also under investigation for use in patients homozygous for Phe508del CFTR mutations.12
References:
- Exclusive Chemistry Ltd. CFTR modulators for treatment of cystic fibrosis-35 compounds. Available at: http://www.exchemistry.com/chem-catalog/cftr-compounds/ Accessed April 20, 2016
- Businesswire.com. Vertex Outlines 2016 Business Priorities to Support the Discovery and Development of New Transformative Medicines for the Treatment of Cystic Fibrosis and Other Serious Diseases. January 10, 2016. Available at: http://files.shareholder.com/downloads/VRTX/0x0x869359/cf7a99bd-bcac-4e8e-bdc9-7f81ac92bccf/VRTX_News_2016_1_10_General.pdf
- Cystic Fibrosis Foundation. Drug Development Pipeline. Available at: https://www.cff.org/trials/pipeline (Accessed April 20, 2016)
- Donaldson S, Pilewski J, Griese M, Dong Q, Lee PS. VX-661, an investigational CFTR corrector, in combination with ivacaftor, a CFTR potentiator, in patients with CF and homozygous for the F508Del-CFTR mutation: interim analysis. J Cyst Fibros. 2013;12:S14.
- Vertex. [press release] Vertex announces data from 12-week phase 2 safety study of VX-661 in combination with ivacaftor in people with cystic fibrosis who have two copies of the F508del mutation. 2016; Available at: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=902790.
- Galapagos NV. Galapagos’ R&D Update 2016. 15 June 2016. Available at: http://www.glpg.com/docs/view/5760e744c26d5-en
- Galapagos NV. Galapagos starts Phase 1 study with potentiator GLPG2451 for CF. 9 May 2016. Available at: https://globenewswire.com/news-release/2016/06/15/848603/10163504/en/Galapagos-R-D-Update-2016.html
- Galapagos NV. Galapagos advances triple combination therapy in cystic fibrosis. Oct. 15, 2015. Available at: https://globenewswire.com/news-release/2015/10/15/776275/10152599/en/Galapagos-advances-triple-combination-therapy-in-cystic-fibrosis.html
- Galapagos NV. Galapagos starts SAPHIRA Phase 2 study with GLPG1837 in cystic fibrosis patients. 16 February 2015. Available at: https://globenewswire.com/news-release/2016/02/16/811175/10160113/en/Galapagos-starts-SAPHIRA-Phase-2-study-with-GLPG1837-in-cystic-fibrosis-patients.html
- Cystic Fibrosis Foundation. Current and Upcoming CFFT Approved Clinical Trials. 2016; Available at: https://CFRD.cff.org/Our-Research/Clinical-Trials/Clinical-Trials-Happening-Now/.
- ClinicalTrials.gov. Study of N91115 in Patients With CF Homozygous for the F508del-CFTR Mutation (SNO-6). 2016; Available at: https://CFRD.clinicaltrials.gov/ct2/show/NCT02589236.
- News Medical. N30 Pharma begins oral dosing in N91115 Phase 1 trial for treatment of cystic fibrosis. 2014; Available at: http://CFRD.news-medical.net/news/20140312/N30-Pharma-begins-oral-dosing-in-N91115-Phase-1-trial-for-treatment-of-cystic-fibrosis.aspx.
Back to top
|
|
KEY TAKEAWAYS
|
|
|
- CFTR modulator therapy can provide important clinical benefit for patients with an appropriate genotype.
- Adherence to therapy with CFTR modulators cannot be assumed and must be considered along with adherence to the usual therapies to achieve best outcomes.
- New drugs are in development, along with new approaches to increase the numbers of patients who will be eligible to start CFTR modulator therapy, but many challenges remain including conducting trials in small numbers of patients with rare genotypes and patients who are very young, very sick, or very well.
Back to top
|
|
IMPORTANT CME/CE INFORMATION
|
|
|
COMPLETE THE
POST-TEST
Click on link to download instructions for the post-test and evaluation
|
|
|
|
ACCREDITATION STATEMENTS
Physicians
This activity has been planned and implemented in accordance with the accreditation requirements and policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint providership of Johns Hopkins University School of Medicine and the Institute for Johns Hopkins Nursing. The Johns Hopkins University School of Medicine is accredited by the ACCME to provide continuing medical education for physicians.
Nurses
The Institute for Johns Hopkins Nursing is accredited as a provider of continuing nursing education by the American Nurses Credentialing Center's Commission on Accreditation.
The Institute for Johns Hopkins Nursing and the American Nurses Credentialing Center do not endorse the use of any commercial products discussed or displayed in conjunction with this educational activity.
CREDIT DESIGNATION STATEMENT
Physicians
eNewsletter: The Johns Hopkins University School of Medicine designates this enduring material for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Nurses
eNewsletter: This 1 contact hour Educational Activity is provided by the Institute for Johns Hopkins Nursing. Each Newsletter carries a maximum of 1 contact hour, or a total of 6 contact hours for the six newsletters in this program.
Respiratory Therapists For United States: Visit this page to confirm that your state will accept the CE Credits gained through this program.
|
|
INTERNET CME/CE POLICY
DISCLAIMER STATEMENT
STATEMENT OF RESPONSIBILITY
STATEMENT OF NEED
CONFIDENTIALITY DISCLAIMER FOR CME ACTIVITY PARTICIPANTS
INTENDED AUDIENCE
HARDWARE & SOFTWARE REQUIREMENTS
|
All rights reserved - The Johns Hopkins University School of Medicine. Copyright 2016.
This activity was developed in collaboration with DKBmed.
|

