 |
|
|
|
|
 |
|

Special Edition: Highlights of the 27th Annual North American Cystic Fibrosis Conference
Welcome to this issue of eCysticFibrosis Review Special Edition, reporting on some of the key information presented at the 27th Annual North American Cystic Fibrosis in Salt Lake City, Utah.
As a special feature, many of these reports include links to podcasts of eCysticFibrosis Review Program Director Peter Mogayzel, MD, discussing the new data with the presenters. Look for the to link to this feature.
to link to this feature.
In this Issue…
The Annual Meeting of the North American Cystic Fibrosis Conference (NACFC) took place this fall in Salt Lake City. The meeting provided unparalleled access to groundbreaking research, new guidelines, and the best in innovative, evidence-based quality education for physicians and health care professionals who treat cystic fibrosis (CF). The conference provides an ideal forum to discuss recent progress in CF research, care, and drug development and to exchange ideas about ways to improve the health and quality of life for people with CF.
This issue looks at new studies that underscore the importance of nutrition in CF health and survival. New evidence-based investigations reveal that lung function and survival in persons with CF are strongly associated with patients achieving a higher body mass index (BMI) at a young age and that CF survival can be quantified as a function of a person�s BMI. We discuss how high doses of dietary fats, while vital for weight gain and growth in CF, may not be good for all people with CF and highlight the effectiveness of behavioral therapies (BEH) in improving a children�s willingness to eat, helping them grow and attain improved health. Other advances beyond nutrition presented at the NACFC included: novel clinical outcomes that are affected by ivacaftor, a CFTR potentiator that targets the basic defect in CF, as part of the first post-approval study in people with CF due to at least one G551D CFTR mutation; an analysis of the CF Foundation Patient Registry that shows the positive effects of long-term use of azithromycin combination therapy against incident nontuberculous mycobacterium (NTM) infections; and a look behind the curtain of the elusive microbiome through lung explant studies.
Among the highlights of the NACFC discussed in this issue are:
| •
| |
CFTR: Ivacaftor improves lung function, lowers sweat chloride levels, and helps patients gain weight. Improved mucociliary clearance (MCC) and sputum rheology reflect a restoration of CFTR activity. |
|
| •
| |
Microbiome: Lung explant sampling differs markedly from standard bacterial cultures in identifying CF lung microbes and colony distributions, as shown in end-stage and non-end-stage tissue samples. |
|
| •
| |
NTM: Patients with chronic P. aeruginosa infection receiving macrolides received the additional benefit of protection against incident NTM infections. |
|
| •
| |
Behavioral and Nutritional Treatment for Preschoolers: Behavioral therapies improve a child�s willingness to eat by teaching caregivers to maximize caloric density, self-monitor, and set goals related to energy intake. |
|
|
| • |
|
High Fat Diets: Too much fat is not right for everyone with CF � diets must be individualized for optimal health. In this study, BMI and lung function show a quantifiable relationship. |
|
| • |
|
Relationship between Nutrition and Survival: Higher weight-for-age percentiles at an early age lead to improved lung function and 18-year survival. |
|
|
|
|
|
|
|
After participating in this activity, the participant will demonstrate the ability to:
 |
|
Discuss the clinical and biological endpoints that reflect effective ivacaftor treatment in people with CF who have at least one G551D CFTR mutation. |
|
|
|
Describe the lung microbiome along with the advantages and disadvantages of different sampling techniques. |
|
|
|
Discuss the role that azithromycin plays in preventing nontuberculous mycobacterium infection and the potential for inducing azithromycin resistance. |
|
|
|
Recognize the advantages of using behavioral therapies to improve the nutritional status of young children with CF. |
|
|
|
Explain the relationship between BMI and FEV1 in underweight and overweight people with CF, and identify those with lower caloric needs. |
|
|
|
Discuss the relationship between nutritional status at a young age and its relationship to growth, health, and survival at puberty and age 18 years. |
|
| The Johns Hopkins University School of Medicine takes responsibility for the content, quality, and scientific integrity of this CME activity. |
|
| |
|
|
| |
 |
| IMPORTANT CME/CE INFORMATION |
 |
|
|
|
|
|
Accreditation Statements
This activity has been planned and implemented in accordance with the Essential Areas and Policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of the Johns Hopkins University School of Medicine and the Institute for Johns Hopkins Nursing. The Johns Hopkins University School of Medicine is accredited by the ACCME to provide continuing medical education for physicians.
The Institute for Johns Hopkins Nursing is accredited as a provider of continuing nursing education by the American Nurses Credentialing Center�s Commission on Accreditation.
The Institute for Johns Hopkins Nursing and the American Nurses Credentialing Center do not endorse the use of any commercial products discussed or displayed in conjunction with this educational activity.
Credit Designations
Physicians
Newsletter:The Johns Hopkins University School of Medicine designates this enduring material for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Nurses
This 1 contact hour Educational Activity is provided by the Institute for Johns Hopkins Nursing. The newsletter carries a maximum of 1 contact hour.
Respiratory Therapists
For United States: Visit this page to confirm that your state will accept the CE Credits gained through this program.
For Canada: Visit this page to confirm that your province will accept the CE Credits gained through this program.
Intended Audience
This activity has been developed for pulmonologists, pediatric pulmonologists, gastroenterologists, pediatricians, infectious disease specialists, respiratory therapists, dieticians, nutritionists, nurses, and physical therapists.
There are no fees or prerequisites for this activity.
Launch Date
This program launched on Jauary 8, 2014 and is published monthly; activities expire two years from the date of publication.
SUCCESSFUL COMPLETION
To successfully complete this activity, participants must read the content, and visit the Johns Hopkins University School of Medicine�s CME website and the Institute for Johns Hopkins Nursing. If you have already registered for other Hopkins CE programs at these sites, simply enter the requested information when prompted. Otherwise, complete the registration form to begin the testing process. A passing grade of 70% or higher on the post-test/evaluation is required to receive CE credit.
Disclaimer Statement
The opinions and recommendations expressed by faculty and other experts whose input is included in this program are their own. This enduring material is produced for educational purposes only. Use of Johns Hopkins University School of Medicine name implies review of educational format design and approach. Please review the complete prescribing information of specific drugs or combination of drugs, including indications, contraindications, warnings and adverse effects before administering pharmacologic therapy to patients.
Statement of Need
Based on a review of the current literature, including national and regional measures, detailed conversations with expert educators at Johns Hopkins, and a survey of potential program participants, this program will address the following core patient care gaps:
| Nutrition |
| • |
Clinicians lack strategies to help patients adhere to CF nutritional requirements, resulting in low body weight and nutritional failure in patients with cystic fibrosis. |
| • |
Clinician uncertainty about how to optimize pancreatic function may lead to ineffective management of pancreatic function in patients with cystic fibrosis. |
|
|
|
|
|
|
|
Planner Disclosure
As a provider approved by the Accreditation Council for Continuing Medical Education (ACCME), it is the policy of the Johns Hopkins University School of Medicine Office of Continuing Medical Education (OCME) to require signed disclosure of the existence of financial relationships with industry from any individual in a position to control the content of a CME activity sponsored by OCME. Members of the Planning Committee are required to disclose all relationships regardless of their relevance to the content of the activity. Faculty are required to disclose only those relationships that are relevant to their specific presentation. The following relationships have been reported for this activity:
Michael P. Boyle, MD, FCCP discloses that he has served as a principal investigator for Vertex, and served on advisory boards for Gilead, Vertex, and Novartis.
No other planners have indicated that they have any financial interests or relationships with a commercial entity.
Guest
Author�s Disclosures
This activity is supported by an educational grant from Aptalis Pharma.
Statement of Responsibility
The Johns Hopkins University School of Medicine takes responsibility for the content, quality and scientific integrity of this CME activity.
Confidentiality Disclaimer for CME Conference Attendees
I certify that I am attending a Johns Hopkins University School of Medicine CME activity for accredited training and/or educational purposes.
I understand that while I am attending in this capacity, I may be exposed to "protected health information," as that term is defined and used in Hopkins policies and in the federal HIPAA privacy regulations (the "Privacy Regulations"). Protected health information is information about a person's health or treatment that identifies the person.
I pledge and agree to use and disclose any of this protected health information only for the training and/or educational purposes of my visit and to keep the information confidential.
I understand that I may direct to the Johns Hopkins Privacy Officer any questions I have about my obligations under this Confidentiality Pledge or under any of the Hopkins policies and procedures and
applicable laws and regulations related to confidentiality. The contact information is: Johns Hopkins Privacy Officer, telephone: 410-735-6509, e-mail: HIPAA@jhmi.edu.
"The Office of Continuing Medical Education at the Johns Hopkins University School of Medicine, as provider of this activity, has relayed information with the CME attendees/participants and certifies that the visitor is attending for training, education and/or observation purposes only."
For CME Questions, please contact the CME Office at (410) 955-2959 or e-mail cmenet@jhmi.edu.
For CME Certificates, please call (410) 502-9634.
Johns Hopkins University School of Medicine
Office of Continuing Medical Education
Turner 20/720 Rutland Avenue
Baltimore, Maryland 21205-2195
Reviewed & Approved by:
General Counsel, Johns Hopkins Medicine (4/1/03)
Updated 4/09
internet
cme/ce policy
The Office of Continuing Medical Education (CME) at the Johns Hopkins University School of Medicine is committed to protecting the privacy of its members and customers. The Johns Hopkins University SOM CME maintains its Internet site as an information resource and service for physicians, other health professionals and the public.
Continuing Medical Education at the Johns Hopkins University School of Medicine will keep your personal and credit information confidential when you participate in a CME Internet-based program. Your information will never be given to anyone outside the Johns Hopkins University School of Medicine�s CME program. CME collects only the information necessary to provide you with the services that you request.
hardware & software requirements
Pentium 800 processor or greater, Windows 98/NT/2000/XP or Mac OS 9/X, Microsoft Internet Explorer 5.5 or later, Windows Media Player 9.0 or later, 128 MB of RAM Monitor settings: High color at 800 x 600 pixels, Sound card and speakers, Adobe Acrobat Reader.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Planning Committee
Michael
P. Boyle, MD, FCCP
Associate Professor of Medicine
Director, Adult Cystic Fibrosis Program
The Johns Hopkins University
Baltimore, Maryland
Peter
J. Mogayzel, Jr., MD, PhD
Professor of Pediatrics
Director, Cystic Fibrosis Center
The Johns Hopkins University
Baltimore, Maryland
Donna W. Peeler, RN, BSN
Pediatric Clinical Coordinator
Cystic Fibrosis Center
The Johns Hopkins University
Baltimore, Maryland
Meghan Ramsay, MS, CRNP
Adult Clinical Coordinator
Cystic Fibrosis Center
The Johns Hopkins University
Baltimore, Maryland |
|
|
|
|
|
|
|
|
|
|
Commentary
& Reviews: |
 |
Peter J. Mogayzel, Jr., MD, PhD
Professor Pediatrics
Director, Cystic Fibrosis Center
The Johns Hopkins University
Baltimore, Maryland
|
|
|
|
 |
|
Stefanie Petrou Binder, MD
Medical Writer
Berlin, Germany |
|
 |
|
Steven Rowe, MD
Associate Professor of Medicine
Division of Pulmonary, Allergy &
Critical Care Medicine
University of Alabama at
Birmingham
Birmingham, Alabama
|
|
 |
|
Scott Powers, PhD, ABPP
Professor of Pediatrics
Director, Center for Child Behavior and Nutrition Research and Training
Cincinnati Children�s Hospital Medical Center
University of Cincinnati College of Medicine
Cincinnati, Ohio |
|
 |
|
Kenneth Olivier, MD, MPH
Principal Investigator
Immunopathogenesis Section
Laboratory of Clinical Infectious Diseases
National Institute of Allergy and Infectious Diseases
Bethesda, Maryland |
|
 |
|
Elizabeth Yen, MD
Assistant Clinical Professor of Pediatrics
University of California at San Francisco
Benioff Children's Hospital
Pleasanton, California |
|
|
Guest Faculty Disclosures
Steven Rowe, MD, has indicated that he has received grant and research funding from Vertex Pharmaceuticals, Inc.
Scott Powers, PhD, ABPP, has indicated that he has received grant and research funding from the NIH and Cystic Fibrosis Foundation.
Kenneth Olivier, MD, MPH, discloses he has received grant and research funding from Insmed, Inc.
No other faculty have indicated that they have any relevant
financial interests or relationships with a commercial
entity.
Unlabeled/Unapproved Uses
Steven Rowe, MD, has indicated that he will discuss off-label and/or investigational uses of ivacaftor.
Kenneth Olivier, MD, MPH, has indicated he will discuss off-label and/or investigational uses of azithromycin, amikacin, tigecycline, imipenem, cefoxitin, linezolid, minocycline, clofazimine, and moxifloxacin.
No other faculty reports that there will be discussion of off-label or investigational uses of any products or drugs.
Planning Committee Disclosures |
|
|
|
|
|
|
|
|
|
|
TO COMPLETE
THE
POST-TEST
Step
1.
Please read the newsletter.
Step
2.
See the post-test link at the end of the newsletter.
Step
3.
Follow the instructions to access the post-test.
|
|
|
|
|
|
|
|
|
|
|
|
|
NACFC Symposium 14.3, Anne Stephenson MD PhD, Division of Respirology,
St Michael’s Hospital, University of Toronto, Adult CF Centre
High Fat Diets in CF: Just Enough or Too Much of a Good Thing? Talk held at the North American Cystic Fibrosis Conference; October 17-19, 2013; Salt Lake City, Utah. |
|
Dietary recommendations for people with cystic fibrosis (CF) must be tailored to each person�s requirements. While excessive fat intake can quickly lead to obesity and health risks in the general population, they form an important part of the CF diet, helping patients increase body weight and improve their survival chances. Nonetheless, the proportion of overweight and obese adults with CF has increased over the past 25 years, as have adverse clinical outcomes such as elevated serum lipids. Although preventing malnutrition continues to be a primary goal of CF care, recent evidence suggests that a lower-calorie diet should be considered in some situations.
Canadian investigator Anne Stephenson MD, PhD, of the Division of Respirology at St. Michael�s Hospital, University of Toronto, who spoke at the North American Cystic Fibrosis Conference, believes that adding fats to the diets of persons with CF has been vital in achieving better health and survival over the past decades.
Survival rates are linked to nutrition, according to a study that focused on US and Canadian patients with CF, between 1972 and 1981. While CF nutritional guidelines at and before the time period studied discouraged dietary fats, patients in Toronto given high fat, high energy diets in the 1970s showed better survival rates than those in the US (Boston), with 50% reaching 30 years of age, compared to 30% reaching 30 years in patients in the US.
Since gaining weight is so important for people with CF, they typically consume a very high fat diet. This diet is key to enabling many children and adults with CF to maintain optimal nutritional status; however, a high fat diet may represent too much of a good thing in some people with CF. A number of studies have shown obesity rates among people with CF ranging roughly from 10%-22%, between 2005 and 2013. Overweight and obese people with CF are generally pancreatic sufficient (PS), older, have higher lung function, and have milder clinical phenotypes. This trend in concerning since it is possible that people with CF will encounter complications from obesity similar to those in the general population.
Dr. Stephenson said that in 2011, 30% of females with CF and 17% of males with CF were classified as underweight (body mass index (BMI) < 20kg/m2), 55% of females and 65% of males had adequate weight (BMI = 20-25 kg/m2), 8% of females and 14% of males were well nourished (BMI = 26-29kg/m2), and 4% of both females and males with CF were obese (BMI ≥ 30kg/m2).
Body mass index trends have changed over time. In a study conducted by Dr. Stephenson consisting of 908 subjects, the adult population was divided into three time periods: before 1990, 1991-1999, and 2000 and later. She found an increase in BMI per year of 3.8% vs 0.4% in PS vs pancreatic insufficient (PI) subjects, respectively. The average BMI before 1990 was 20.7 � 2.7 kg/m2, while the average BMI after 2000 was 22.3 � 3.4 kg/m2.
Dr. Stephenson found a quantifiable relationship between BMI and lung function in people with CF. In an investigation that included 426 individuals with CF (68 underweight, 288 adequate, and 70 obese), she noted a 10% increase in BMI was associated with a 4% increase in FEV1 in those who were underweight. By contrast, in those who were overweight, a 10% increase in BMI was associated with a 2% increase in FEV1. Furthermore, a 10% increase in BMI was associated with a 4.3% increase in FEV1 in patients who were PI. In patients who were PS, a 10% increase in BMI was associated with a 2.7% increase in FEV1.
Overall, the better nourished individuals with CF are, the better their FEV1 and survival. A high fat diet is recommended for those with CF, since improvements in BMI translate into improvements in lung function. However, there is small benefit in this in those whose BMI exceeds 23-25 kg/m2 in whom dietary intake should be balanced against the known health risks for obesity.
|
|
| back
to top |
|
|
|
|
|
|
|
|
|
|
|
Nontuberculous Mycobacterium |
|
|
|
|
| Abstract 271: Epidemiology of Nontuberculous Mycobacterial Infection and Associated Macrolide Use Among Persons with Cystic Fibrosis. Binder A, Adjemian J, Olivier KN, Prevots DR. Abstract from the North American Cystic Fibrosis Conference. October 17-19, 2013, Salt Lake City, Utah. |
|
The survival of patients with cystic fibrosis (CF) has been threatened by the development of highly resistant bacterial strains. Joining the growing list of resistant pathogens, which most notably includes Pseudomonas aeruginosa, Staphylococcus aureus, and Burkholderia cepacia, among others, are nontuberculous mycobacteria (NTM), which are largely ubiquitous, free-living inhabitants of the natural environment. NTM infection causes progressive lung damage in people with CF and is playing an increasingly recognized role in CF lung disease. The potential risk of mycobacterial resistance to antibiotic treatment makes prompt identification of newly acquired NTM cases a firm priority.
According to Alison Binder, MS, of the Epidemiology Unit of the National Institutes of Health in Bethesda, Maryland, people with CF are at a high risk of NTM infection, and its treatment requires prolonged combination drug regimens that include macrolides, such as azithromycin. However, azithromycin is also currently used to manage people with CF who have chronic P. aeruginosa infections and are not infected with NTM, which raises growing concerns about the development of macrolide-resistant NTM strains.
In her presentation at a workshop on Epidemiology & Management of CF Pulmonary Infections at the North American Cystic Fibrosis Conference, Ms. Binder discussed the relationship between macrolide use and NTM infections among people with CF, revealing that azithromycin therapy actually protected against incident NTM infections. Nevertheless, with the growing concerns about macrolide resistance with azithromycin monotherapy in occult NTM infection, annual screening for NTM should be performed in people with CF who are receiving azithromycin therapy. Using patient data from the 2011 CF Foundation Patient Registry, Ms. Binder and her team conducted a nested case-controlled study among people with CF who had completed a mycobacterial culture in 2011. She defined incident NTM cases as those who had a positive culture in 2011 only, with no positive cultures reported in 2010, or cases of NTM disease reported in the data from 2003-2010 (culture data was not available before 2010). The study defined controls as those with negative cultures in 2010-2011 and no prior reported NTM. Those who died in 2011, were missing annual data, or received transplants in any year were excluded. Associations between past macrolide use and incident NTM infections were evaluated using odds ratios from logistic regression.
The 2011 CF Foundation Patient Registry included 27,112 patients, of whom 18,771 (69%) were cultured for mycobacteria in 2010-2011. Of those, 956 (5%) had new NTM in 2011, including 566 (59%) with Mycobacterium avium complex (MAC) and 326 (34%) with Mycobacterium abscessus. The study included 16,168 controls.
Thirty-three percent of the MAC and M. abscessus cases involved treatment with azithromycin in 2010, compared with 50% of the controls. Both the MAC and M. abscessus case groups were significantly less likely than controls to report azithromycin use in 2010 (OR [odds ratio] = 0.5, P < 0.001 for both). This association varied by age: a protective association was observed among adolescents aged 12-17 years (OR = 0.4, P < 0.001) and adults aged 18 or older (OR = 0.5, P < 0.001), but not among children between 5-12 years (OR = 0.7, P = 0.1). In the data obtained for 2003-2011, a significant dose response effect was observed among adolescent and adult patients who, because of the increasing duration of macrolide use, showed an increased protection against incident MAC and M. abscessus infection.
|
|
| back
to top |
|
|
|
|
|
|
|
|
|
|
|
|
Weight – Height – Lung Volume – Survival |
|
|
|
|
| NACFC Symposium14.2, Elizabeth Yen, MD, Assistant Clinical Professor of Pediatrics, University of California, San Francisco; UCSF Benioff Children�s Hospital |
|
Nutrition is a strong predictor of morbidity and survival in cystic fibrosis (CF). A new US study maintains that greater weight gain at four years of age is associated with greater height, better pulmonary function, fewer CF complications, and better survival through age 18. Furthermore, improved weight-for-age in the peripubertal period can help promote an improved tempo and timing of pubertal height growth. Taking an in-depth look at the effects of nutrition in CF at the North American Cystic Fibrosis Conference was Elizabeth Yen MD, Assistant Clinical Professor of Pediatrics, University of California, San Francisco; Benioff Children's Hospital.
Dr. Yen conducted a retrospective study using data from the CF Foundation Patient Registry for individuals born between 1989 and 1992 (N = 3142). She evaluated the impact of nutritional status early in life on the timing and velocity of height, growth, lung function, survival, and CF complications through age 18 (Table).
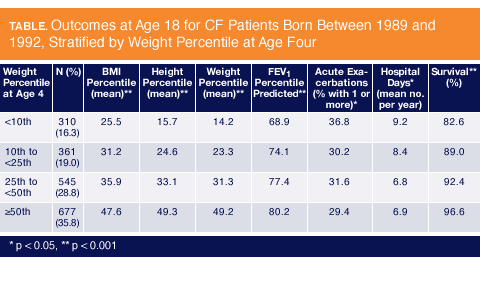 Individuals with CF were stratified according to their weight-for-age percentile (WAP) at age 4-5 years (< 10%, 10-25%, 25-50%, >= 50%), and the study endpoints were assessed through 2009. The mean height velocity was calculated as a moving average for every 0.2 year of age.
Individuals with CF were stratified according to their weight-for-age percentile (WAP) at age 4-5 years (< 10%, 10-25%, 25-50%, >= 50%), and the study endpoints were assessed through 2009. The mean height velocity was calculated as a moving average for every 0.2 year of age.
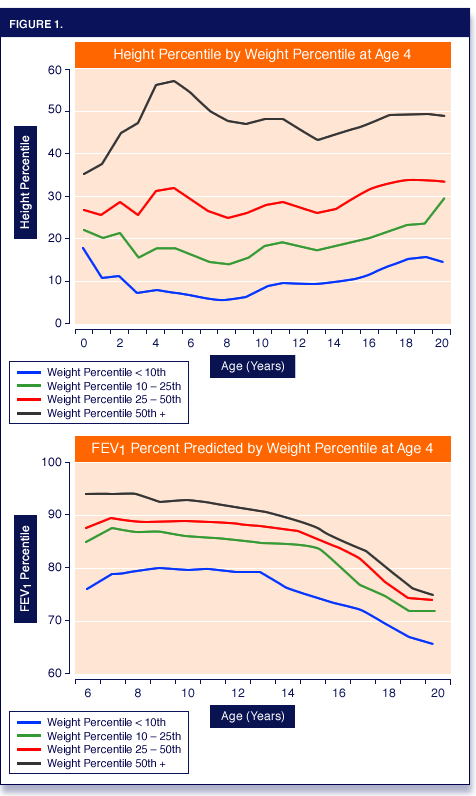
The WAP at 4 years of age was positively associated with height-for-age percentiles (HAP) throughout childhood. At the age of 4 years, a WAP > 10% was associated with better lung function between the ages of 6-18 years, as well as with progressively better outcomes in all categories measured (Figure 1).
 By the age of 18, children who fell short of the 10th WAP percentile had gained less weight and grew less in height than children with better weight-for-age percentiles.
They achieved substantially lower mean body mass indices (BMI) and had poorer lung function, as reflected by FEV1 percent predicted, than better-nourished children. Conversely, 18 year olds who achieved higher WAP percentiles as children (10-25%, 25-50%, and >= 50%) had better outcomes for each endpoint than the groups below them.
By the age of 18, children who fell short of the 10th WAP percentile had gained less weight and grew less in height than children with better weight-for-age percentiles.
They achieved substantially lower mean body mass indices (BMI) and had poorer lung function, as reflected by FEV1 percent predicted, than better-nourished children. Conversely, 18 year olds who achieved higher WAP percentiles as children (10-25%, 25-50%, and >= 50%) had better outcomes for each endpoint than the groups below them.
Dr. Yen�s analysis shows that the number of acute exacerbations per year was directly related to WAP in these individuals. The percentage of 18 year olds experiencing one acute exacerbation or more was highest in children who were categorized below the 10th percentile and lowest in children categorized above the 50th percentile. The same pattern was observed for the length of stay in hospital per year, with better-nourished children spending less time in hospital than less adequately nourished children.
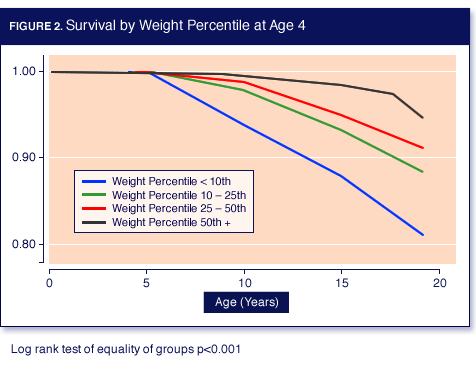
Better survival at the age of 18 years was strongly related to higher weight-for-age-percentiles in children. Dr. Yen revealed a survival rate at 18 years of 82.6% in children stratified into the lowest percentile group. The survival rate rose steadily in each progressive WAP group, revealing a 96.6% survival rate in children with WAP >= 50% (Figure 2).
In boys and girls with current WAP > 50%, peak pubertal height velocities approximated but remained lower than that of the healthy reference population. Patients attaining higher WAP and height-for-age percentiles at 4 years had a survival advantage throughout childhood.
|
|
| back
to top |
|
|
|
|
|
|
|
|
|
|
|
Microbiome Studies: Lung Sections Tell |
|
|
|
|
NACFC Symposium 4.1, Lucas Hoffman, MD, PhD. Associate Professor, Pediatrics and Microbiology, University of Washington
CF Lung Microbiome: What Have We Learned? Lucas Hoffman, MD, PhD. Presentation at the North American Cystic Fibrosis Conference, October 17-19, 2013; Salt Lake City, Utah. |
|
The structure and dynamics of bacterial communities in the airways of people with cystic fibrosis (CF) remain largely unknown, and few studies have shed light on the complexity of the CF lung microbiome. Recent evidence from explanted lung sections suggests that standard cultures do not accurately reflect the microbiome diversity.
Studies to date have focused largely on microbes detected in CF secretions, as identified through sputum, oropharyngeal (OP) swabs, and bronchoalveolar lavage fluid. While relatively easy for the clinician to obtain, some evidence suggests that such samples may be contaminated by oral cavity and upper airway flora and are therefore a poor reflection of actual lung colonization. Investigations using direct sampling of lung explant sections and postmortem lung tissue may provide a more specific characterization of the microbiota in the CF lung, than standard sampling-methods.
Leading a discussion on the subject of the CF Lung Microbiome: What Have We Learned? at the North American Cystic Fibrosis Conference, Dr. Lucas Hoffman, MD, Associate Professor of Pediatrics and Microbiology at the University of Washington, highlighted current, relevant investigations in lung explant studies.
In one such study, the diversity of microbial communities in tissue sections from anatomically distinct regions of the CF lung was determined using 16S rDNA gene amplicon pyrosequencing. Microbial communities differed significantly between different areas of the lungs, indicating that CF infections are not only polymicrobial but are also spatially heterogeneous. This suggests that treatments that target microbes identified in sputum or BAL samples may be ineffective against infections in some areas of the lung. According to the study, microbial community richness was greater in the explant lungs than in the postmortem lungs.
Similar findings were recorded in an investigation that compared end-stage and non-end-stage lungs, using standard culture techniques and molecular methods. Tissue and sputum samples were obtained from the explanted lungs of 5 individuals with end-stage CF lung disease that were examined along with routine expectorates from patients with non-end-stage CF, representing earlier stages of chronic lung disease. Pseudomonas aeruginosa was the sole pathogen in end-stage CF lungs. Conversely patients with non-end-stage CF were found to harbor several species, which were unevenly distributed across the lung, with microbes found in monospecies aggregates. The outcomes indicated that standard culture technique could identify only the dominant pathogens.
Conflicting outcomes and opinions mark the subject of the microbiome sampling procedures. In a third study involving lung explants, investigators contended that sampling lung secretions through the oropharynx could contaminate the results. A comparison of culture-independent analyses of throat and sputum specimens with samples from lung transplants showed relatively homogeneous populations of typical CF pathogens in the lung samples of patients with advanced disease.
In contrast, upper airway specimens from the same individuals had a higher microbial diversity, including some organisms that are not considered CF pathogens; this underscores the importance of sampling procedures for microbiome studies.
In spite of the elucidating results obtained from explant and postmortem tissue analyses, it can be argued that the specimens reflect the microbiota found in end-stage disease and consequently are of no use to the clinician. Dr. Hoffman shared the results of an as-yet unpublished study using data obtained from a lung lobe sample of a child with CF. The findings corroborate the explant study observation that OP swabs or culture reveal markedly different microbes than serial lobe sections. The study further corroborated the explant studies in showing that the CF lung microbial communities were spatially fairly heterogeneous and were characterized by an abundance of anaerobic and barely identified species.
Dr. Hoffman noted that the high interpatient variability of the species composing the microbiome limits our ability to apply information learned about one patient to another. The biases, sources of error, and disadvantages conveyed by the different sampling techniques and in the absence of the means to correlate results with clinical outcomes led Dr. Hoffman to call for further research for consistent, clinically useful predictors of exacerbation onset, clinical course, and response to treatment. |
|
| back
to top |
|
|
|
|
|
|
|
|
|
|
|
The Goal Study – Ivacaftor Use In Real Life |
|
|
|
|
Abstract 206: Results of the G551D Observational Study: The Effect of Ivacaftor in G551D Patients Following FDA Approval; Rowe SM, Heltshe SL, Gonska T, Donaldson S, Borowitz D, Gelfond D, Sagel SD, Khan U, Hamblett NM, VanDalfsen J, Joseloff E, Ramsey B. Abstract from the North American Cystic Fibrosis Conference; October 17-19, 2013; Salt Lake City, Utah.
Abstract 209: Effect of Ivacaftor on Mucociliary Clearance and Mucus Rheology in Patients with a G551D CFTR Mutation; Donaldson SH, Zeman K, Laube B, Corcoran T, Locke LW, Pilewski J, Hanes J, Schuster B, Kanzawa M, Rowe SM, Bennett WD. Abstract from the North American Cystic Fibrosis Conference; October 17-19, 2013; Salt Lake City, Utah. |
|
Ivacaftor is a drug that has stirred excitement in the cystic fibrosis (CF) field because it targets the underlying cause of CF and does not just address its various symptoms. Ivacaftor improves lung function, lowers sweat chloride levels, and ultimately helps patients gain weight, which is crucial for well-being in people with CF who have at least one G551D CFTR mutation.
The effects of ivacaftor in a post-approval setting were shown by the recent results of the G551D Observational (GOAL) Study. Steve Rowe, MD, MSPH, University of Alabama, Birmingham, speaking at the North American Cystic Fibrosis Conference on behalf of the GOAL investigators, confirmed that ivacaftor is a CFTR potentiator with profound benefits in people with cystic fibrosis caused by at least one G551D CFTR mutation.
The prospective, multicenter, large observational cohort study that included several substudies, recruited patients of at least 6 years of age with a confirmed diagnosis of CF based on positive sweat chloride ≥ 60 mEq/liter (by pilocarpine iontophoresis) and /or a genotype with two identifiable mutations consistent with CF, at least one copy of the G551D CFTR mutation, and no prior exposure to ivacaftor. One hundred fifty-one individuals were included (38 age 6 to 11 years and 113 age ≥ 12 years).
Eighty-eight percent of the study participants (133/151) completed the six-month study period. The average patient age was 21.1 � 11.4 years; 46.4% were female, and 72.2% were heterozygous for F508del.
After six months of ivacaftor therapy, lung function improved significantly from the mean baseline FEV1% predicted of 82.6 �25.6, with a mean change of 6.7% (95% CI [confidence interval] = (4.9, 8.5), P < 0.0001) and was detectable at the first (one month) follow-up visit (mean change = 6.7%, 95% CI = (5.2, 8.3), P < 0.0001). Similar improvement was seen in FVC % predicted.
Sweat chloride was reduced from baseline (102.9 � 13.8 mEq/L) by a mean change of -53.8 � 22.2 (95% CI = (-57.7, -49.9), P < 0.0001) at 6 months, which confirmed that ivacaftor was activating chloride transport and restoring CFTR function. The mean sweat chloride at one month was 55.2 � 23.6 mEq/L, reflecting ~30% CFTR function.
The mean change seen in body weight by six months from baseline was 2.5 kg (95% CI = (1.9, 3.1), P < 0.0001). Furthermore, there was significant improvement in multiple quality of life measures.
The GOAL study investigated a broad range of clinical and biological endpoints that reflect restoration of CFTR activity in patients with the G551D mutation. In one GOAL substudy, mucociliary clearance (MCC) and sputum rheology outcomes proved that restoration of CFTR activity significantly increased MCC and improved mucus rheological properties.
Scott Donaldson MD, Associate Professor at the University of North Carolina, Chapel Hill spoke about the Effect of Ivacaftor on Mucociliary Clearance and Mucus Rheology in Patients with a G551D CFTR Mutation, as part of a GOAL substudy that included patients with CF patients older than 6 years who had at least one G551D mutant allele and an FEV1 > 30% predicted. The study participants discontinued hypertonic saline and rhDNase treatment before each study visit (12 and 24 hours, respectively).
Twenty-one subjects completed all pre- and posttreatment MCC scans. At one month after ivacaftor initiation, mucociliary clearance (AUC60) increased from 8.5 � 1.7% at baseline to 18.7 � 2.3% and 17.7 � 1.7% at one and three months posttreatment, respectively (P < 0.001 for each comparison to baseline). Dramatic improvements in peripheral lung clearance were also demonstrated (1.5 � 1.8% vs. 12.2 � 2.0%, 9.1 � 2.4% at baseline, one month, and three months, respectively; P < 0.05 for each comparison to baseline), which is where CF lung disease and delayed MCC are most pronounced. Interestingly, the improvement in lung function and MCC corresponded with a decrease in the rate of P. aeruginosa infection, which decreased from 52% before initiation of therapy to 34% of those taking ivacaftor after six months of therapy (P < 0.01 by Wilcoxon sign test).
|
|
| back
to top |
|
|
|
|
|
|
|
|
|
|
|
Behavioral Therapies In CF Nutrition |
|
|
|
|
| Abstract 532: A Multi-site, Randomized, Controlled Clinical Trial of Behavioral and Nutrition Treatment for Preschoolers with Cystic Fibrosis. Powers SW, Stark L, Chamberlin L, Sullivan S, Filigno S, Rausch J. Abstract from the North American Cystic Fibrosis Conference; October 17-19, 2013; Salt Lake City, Utah. |
|
Early treatment for suboptimal growth is a critical target for intervention in children with cystic fibrosis (CF), because of evidence that sustained weight gain at a young age can help improve lung function, limit lung structural damage, and reduce morbidity over time. Nevertheless, children with CF frequently fail to meet nutritional recommendations.
Nutrition interventions that incorporate behavioral therapies (BEH) show more promise in increasing energy intake in children with CF than attention control nutritional interventions alone (CTL), according to the results of a trial that investigated BEH versus CTL in preschool children with CF and pancreatic insufficiency. Behavioral therapies incorporate factors such as mealtime duration, family mealtime interaction, and the child�s mealtime behavior to achieve higher caloric intake. Attention control nutritional interventions provide education and guidance for patient families, serving more as a reminder for attention control and contact frequency.
According to Scott Powers PhD, ABPP, who presented the trial at the North American Cystic Fibrosis Conference as part of a workshop titled Current Topics in Nutrition, BEH was safe, well tolerated by young children, and effective for improving energy intake and height for age z-scores. Furthermore, the study supported the current CF Nutrition Clinical Care Guidelines and seemed to indicate that a greater energy intake could help make a clinical impact on weight in this age group.
The multisite, randomized, controlled clinical trial included 78 young children with CF between the ages of two and six years with pancreatic insufficiency who came from seven CF centers and their families. The patients were randomized to either BEH (n = 36) or CTL (n = 42). The groups were stratified according to baseline weight-for-age (WFA) z-score (Strata: <= -1.0 and > -1.0 to +1.0).
At baseline, the mean age of the children was 3.8 � 1.3 years; 55% were female and 45% were male. The baseline energy intake was 1462 � 330 calories. The mean baseline WFA z-score was - 0.44 � 0.81, and the height-for-age (HFA) z-score was -0.55 � 0.84. Airway cultures from 5% of the children grew Pseudomonas aeruginosa, and no significant differences were noted between the groups at baseline.
Fifty-three percent received BEH or CTL via face-to-face delivery, while the remainder had telehealth treatment delivery. Treatments occurred weekly for eight weeks and then monthly for four months, after which the participating patients returned to standard care, followed by a 12-month follow-up assessment. The three primary aims of the study were to test the daily energy intake change in patients (measured by seven-day, weighed diet diaries), weight for age z-score changes from baseline to post-treatment (six months), and height for age z-score changes from baseline to follow-up (18 months).
Behavioral therapies produced greater changes than CTL in energy intake (difference in change: +431 kcal/day; P < 0.001) and HFA z-score (difference in change: + 0.14 cm/12 months; P < 0.05), while the WFA z-score change (difference in change: + 0.09 kg/12 months; P = 0.25) was not significant. Few adverse events were reported, and retention of participants was excellent.
Dr. Powers noted that meeting CF care nutritional goals for preschoolers is challenging because of developmental, psychosocial, and medical factors. Behavioral therapies help caregivers learn how to maximize caloric density in food, self-monitor, and set clear goals for energy intake. BEH teaches principles from behavioral therapy that increase a child�s motivation to eat and help address mealtime behavior challenges typical of young children. Improving outcomes for both energy intake and growth are critical targets for CF care and clinical research. Early interventions are needed to optimize these outcomes.
|
|
| back
to top |
|
|
|
|
|
|
COMPLETE
THE
POST-TEST
Step
1.
Click on the appropriate link below. This will take you to
the
post-test.
Step
2.
If you have participated in a Johns Hopkins on-line course,
login. Otherwise, please register.
Step
3.
Complete the post-test and course evaluation.
Step
4.
Print out your certificate.


* (The post-test for the newsletter is 1 credit hour.)
Respiratory
Therapists
Visit
this page to confirm that your state will accept the
CE Credits gained through this program or click on the
link below to go directly to the post-test.
 |
|
|
|
|
|
|
|
|
|
|
|
|
© 2014 JHUSOM, IJHN and eCysticFibrosis Review
Presented by JHUSOM and IJHN in collaboration with DKBmed. |
|
|
|
|
|
|
|
|
Illustration
� Michael Linkinhoker |
|
|
|
|
|







